
第1题:
参考答案:
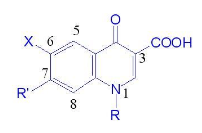
(1)N1位取代基可为脂肪基和芳烃。
(2)2-位引入取代基后,其活性减弱或消失。
(3)3位羧基和4位酮基是该类药物与DNA回旋酶结合产生药效必不可少的部分。
(4)5位取代基中,氨基取代时活性最好,其他基团取代活性均降低。
(5)6位取代基的活性大小顺序位F>Cl>CNNH2>H。
(6)7位引入不同取代基均可提高抗菌活性,对活性贡献顺序哌嗪>N(CH3)2>CH3>Cl>H。
(7)8位的取代基可以使H、Cl、F、NO2、NH2等,以F为最佳。
第2题:
噻嗪类利尿剂和哪类降压药物合用可减少发生低血钾的可能性()。
第3题:
利尿剂、长效二氢吡啶类钙拮抗剂( )
第4题:
用做抗高血压的二氢吡啶类拮抗剂的构效关系研究表明()
第5题:
简述ACEI的构效关系。
第6题:
二氢吡啶类钙拮抗剂的的相对禁忌证包括()和()。
第7题:
简述镇痛药物的构效关系。
第8题:
第9题:
简述二氢吡啶类钙拮抗剂的构效关系。
第10题:
列举三种整合酶抑制剂,并简述其构效关系特点。